
Epidermolysis Bullosa (EB), czyli pęcherzowe oddzielanie się naskórka, jest grupą genetycznie uwarunkowanych chorób pęcherzowych skóry charakteryzujących się powstawaniem pęcherzy samoistnie lub po niewielkim urazie mechanicznym. Częstość choroby szacuje się na 1/50 000 żywych urodzeń.
Spis treści:
1. Objawy
2. Dziedziczenie
3. Klasyfikacja
1. Objawy
Główne objawy kliniczne związane z EB spowodowane są mechaniczną wrażliwością skóry. Stąd tendencja do powstawania zmian skórnych, do których w pierwszym rzędzie należy zaliczyć pęcherze, prosaki czyli milie, przebarwienia pigmentacyjne, nadżerki i blizny, ubytki naskórka, włączając w to wrodzony brak naskórka, zmiany w obrębie płytek paznokciowych obejmujące zarówno dystrofię, jak i brak paznokci, łysienie. Tworzenie się pęcherzy i blizn w okolicach palców dłoni i stóp prowadzi do ich przykurczów i zrostów (tzw. pseudosyndaktylia). Bliznowaceniu może towarzyszyć dotkliwy świąd. Objawy skórne nie są jedyną konsekwencją EB. Zmiany pojawiają się także wewnątrz ciała powodując powstanie pęcherzy, nadżerek, blizn, zrostów prowadzących do przewężeń także i w obrębie przewodu pokarmowego, dróg moczowych, płuc.
Epidermolysis Bullosa jest grupą chorób, które różnią się od siebie przebiegiem klinicznym, stąd występowanie, nasilenie i lokalizacja objawów jest u pacjentów zróżnicowana. U niektórych pacjentów zmiany skórne mogą być ograniczone do pojedynczych, gojących się zmian pęcherzowych, podczas gdy u innych mogą występować przewlekle obejmując obszar całego ciała.
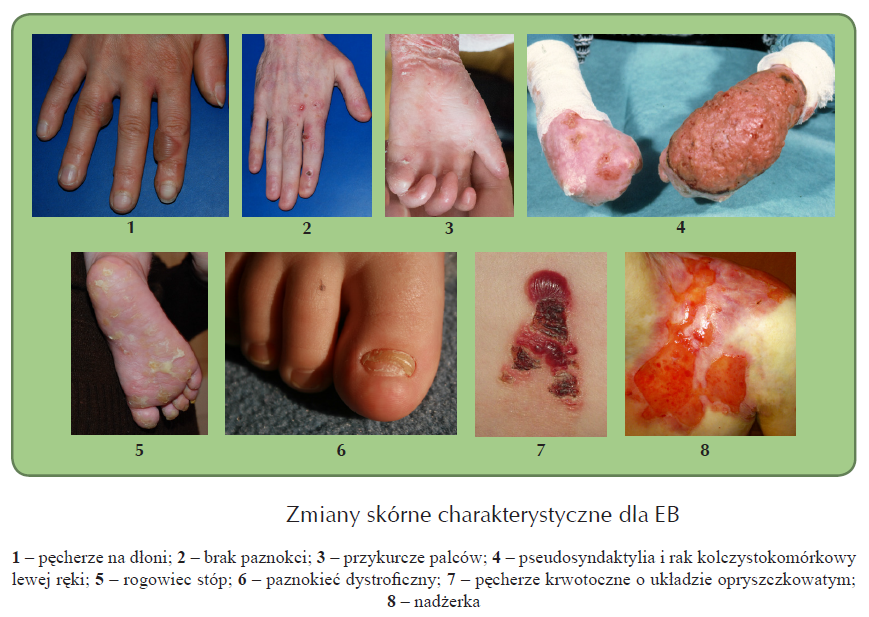
Zróżnicowany przebieg kliniczny EB
Choroba jest wynikiem nieprawidłowego połączenia naskórka i skóry właściwej. Naskórek jest najbardziej zewnętrzną częścią powłoki skórnej i bierze udział w ochronie organizmu przed działaniem czynników zewnętrznych, w tym m.in. promieniowania UV, patogenów, a także zapobiega utracie wody. Leżąca poniżej skóra właściwa jest miejscem, gdzie znajdują się naczynia krwionośne, jak również zakończenia nerwowe. W warunkach prawidłowych połączenie pomiędzy tymi warstwami jest ścisłe, elastyczne i wytrzymałe na urazy mechaniczne. Połączenie to zbudowane jest z kilkunastu cząsteczek białkowych. Cząsteczki te ściśle ze sobą współdziałają, dlatego brak lub nieprawidłowa budowa którejkolwiek z nich powoduje osłabienie połączenia naskórka i skóry właściwej.
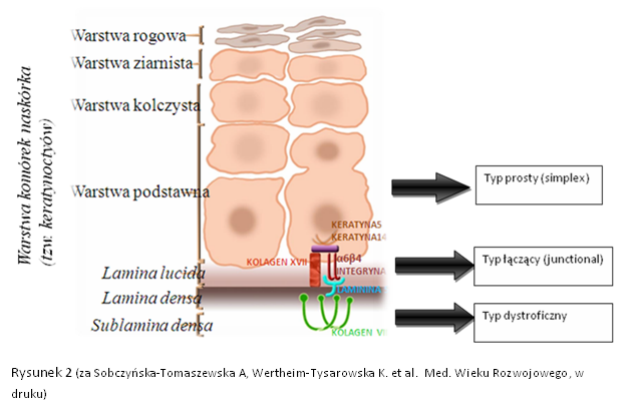
Poniższe ryciny 1 i 2 przedstawiają przekrój zdrowego naskórka oraz skóry właściwej z zaznaczonymi białkami strukturalnymi łączącymi warstwy skóry.
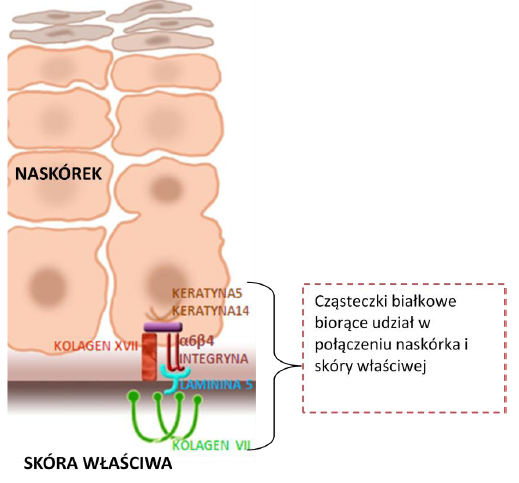
Ryc. 1
Ryc. 2
2. Dziedziczenie
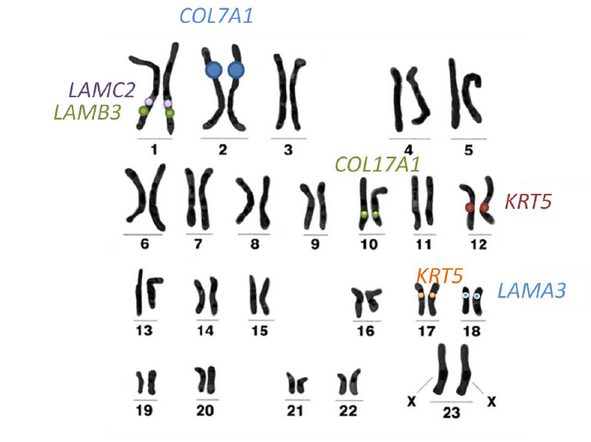
Ryc. 3
U każdego człowieka w każdej komórce jądrzastej (czyli w większości komórek ciała) występuje 46 chromosomów, spośród których dwa to chromosomy płci (u kobiet XX, u mężczyzn XY). Na 44 pozostałe chromosomy składają się 22 pary chromosomów. Z danej pary chromosomów jeden jest dziedziczony od matki, a drugi od ojca. W ten sposób każdy z nas ma dwie kopie każdego genu – jedną odziedziczoną od matki, drugą od ojca (z wyjątkiem tych znajdujących się na chromosomie X i Y, ale chromosomy X i Y nie są związane z EB). Na rycinie 3 przedstawiono przykładowy zestaw chromosomów człowieka (tzw. kariotyp) z zaznaczoną lokalizacją wybranych genów odpowiedzialnych za EB.
Zależnie od typu i podtypu EB oraz tego, w którym genie wystąpiła mutacja, a także rodzaju mutacji i jej lokalizacji w genie, choroba może być dziedziczona w sposób autosomalny recesywny lub autosomalny dominujący.
Dziedziczenie autosomalne recesywne (Schemat 1A) oznacza, że objawy choroby występują wtedy, gdy obie kopie danego genu zawierają mutacje. U osób, które mają jedną kopię genu z mutacją, a drugą prawidłową nie występują objawy choroby – osoby takie nazywa się nosicielami (bo są nosicielami mutacji w danym genie). Tak więc w tym przypadku u rodziców chorych dzieci mogą nie występować objawy choroby, ale jeśli dziecko odziedziczy od każdego z rodziców kopię genu z mutacją, wówczas u niego wystąpią objawy EB.
Dziedziczenie autosomalne dominujące (Schemat 1B) oznacza, że wystarcza tylko jedna kopia zmutowanego genu, aby wystąpiły objawy choroby. Oznacza to, że jeśli rodzic jest chory na taką postać EB, to jeśli przekaże swojemu dziecku kopię genu z mutacją, ono też będzie chore.
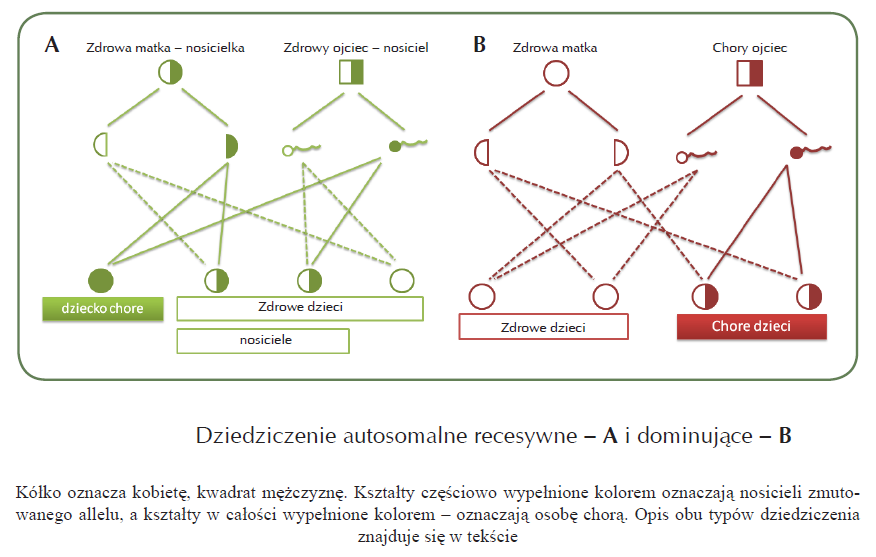
Schemat 1
Większość przypadków typu prostego (simplex) jest dziedziczona w sposób dominujący, a mutacje dotyczą genów KRT5 i KRT14. Znacznie rzadziej – około 5% przypadków EB typu prostego (simplex) to choroba dziedziczona autosomalnie recesywnie i spowodowana mutacjami w genie KRT14. Postaci EB typu prostego (simplex) spowodowane mutacjami w innych genach (ITGA6, ITGB4, DSP1, PKP1) są dziedziczone w sposób autosomalny recesywny. Wyjątek stanowi gen PLEC1, którego mutacje mogą występować zarówno w dziedziczonym w sposób autosomalny dominujący, jak i recesywny podtypie EB typu prostego (simplex).
W przypadku typu łączącego (junctional) do roku 2009 uznawano, iż wszystkie postaci spowodowane mutacjami w genach LAMB3, LAMC2, LAMA3, COL17A1, ITGA6 lub ITGB4 dziedziczone są w sposób autosomalny recesywny. W 2009 roku opisano pierwszy na świecie przypadek 7-letniej dziewczynki, u której stwierdzono dominującą postać EB typu łączącego (junctional) spowodowaną mutacją w genie COL17A1.
W przeciwieństwie do pozostałych typów choroby, typ dystroficzny jest powodowany mutacjami w jednym tylko genie – COL7A1. Mutacje w tym genie, zależnie od rodzaju i lokalizacji, mogą odpowiadać zarówno za autosomalny dominujący, jak i autosomalny recesywny typ dziedziczenia choroby.
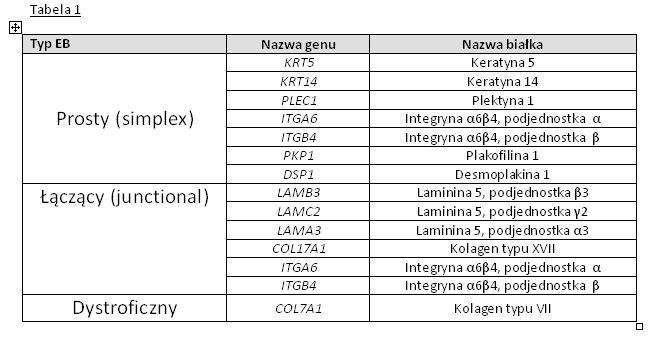
Tabela 1: Zestawienie genów kodujących odpowiednie białka w skórze oraz typy EB zależne od uszkodzenia danego genu
3. Klasyfikacja
Klasyfikacja EB uwzględnia trzy główne typy choroby: prosty (simplex), łączący (junctional) i dystroficzny. Każdy z tych typów jest spowodowany nieprawidłowościami w różnych cząsteczkach białkowych i charakteryzuje się różnym przebiegiem klinicznym.
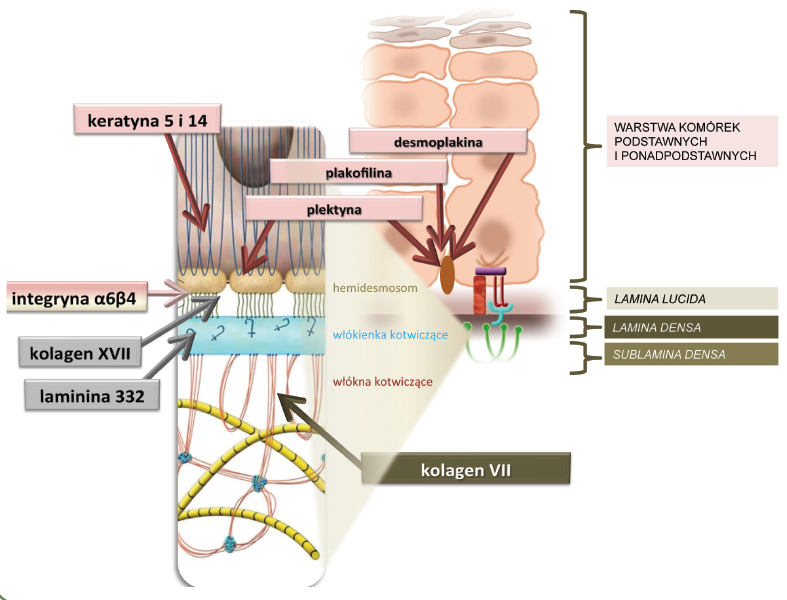
Podstawą do wyróżnienia trzech typów EB jest badanie mikroskopowe wycinka skóry. Celem badania jest określenie miejsca w przekroju granicy naskórka i skóry właściwej, w którym powstaje pęcherz. Na granicy tej wyróżnia się kilka warstw widocznych w mikroskopie. Jeśli pęcherz u pacjenta powstaje w warstwie komórek naskórka, wówczas rozpoznaje się typ prosty (simplex), jeśli w obrębie tzw. lamina lucida, rozpoznaje się typ łączący (junctional), a jeśli w warstwie zwanej sublamina densa, rozpoznaje się typ dystroficzny (Ryc. 4 i 5).
Ryc. 4
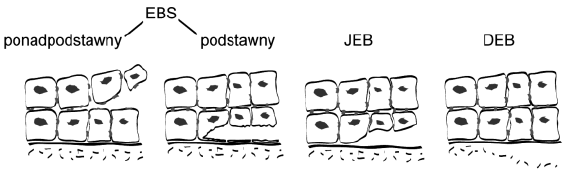
Ryc. 5
Każdy z trzech wyróżnianych typów EB różni się przebiegiem klinicznym, jednak w obrębie każdego z nich występują zarówno postaci o łagodniejszym, jak i bardzo ciężkim przebiegu klinicznym (Schemat 2).
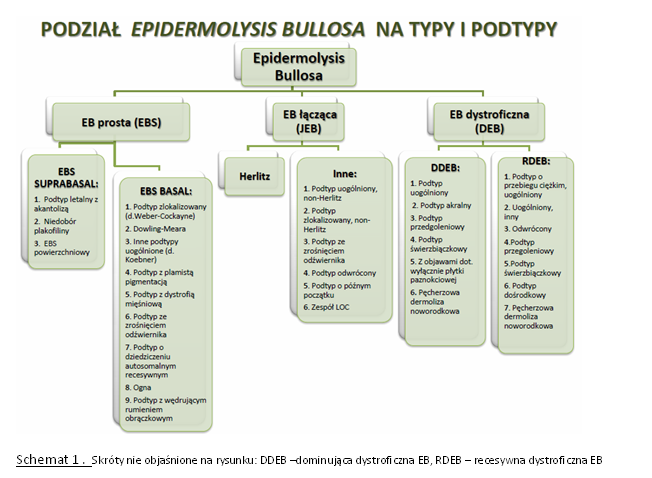
Schemat 2: Podział typów EB
Artykuł przygotowany z wykorzystaniem:
[1] skryptu “Epidermolysis bullosa – pęcherzowe oddzielanie się naskórka. Etiopatogeneza, dziedziczenie, diagnostyka, leczenie.” Katarzyna Wertheim-Tysarowska, Cezary Kowalewski, Katarzyna Woźniak, Jerzy Bal, Wyd. Continuo;
[2] publikacji “GENETYCZNIE UWARUNKOWANE CHOROBY SKÓRY – PRZEGLĄD WYBRANYCH GENODERMATOZ”, Medycyna Wieku Rozwojowego, 2012, XVI, 3, © IMiD, Wydawnictwo Aluna.






